Galaxy Australia is capable of conducting genome annotation using the FgenesH++ annotation tool. Users need to apply for access to this tool, please see service notes here and apply for access here.
This How-to-Guide will describe the steps required to annotate your genome on the Galaxy Australia platform (see Fig 1), developed in consultations between the Bioplatforms Australia Threatened Species Initiative, Galaxy Australia, and Australian BioCommons.
If you need help, the Galaxy community is both approachable and helpful. Ask them questions!
Quick start guide
- Log in to Galaxy Australia
- Apply for access to FGenesH++.
- Create a new history
- Upload your
assembled reference genome,repeat masked reference genome,.cdna,.proand.datfiles from the transcriptome workflow - Load and execute workflows, using required options
- Review workflow report and perform additional QC as needed
- Re-run workflows, or individual tools, as needed
How to cite the workflow
Silver, L. (2024). Fgenesh annotation -TSI. WorkflowHub. https://doi.org/10.48546/WORKFLOWHUB.WORKFLOW.881.5
The overall workflow
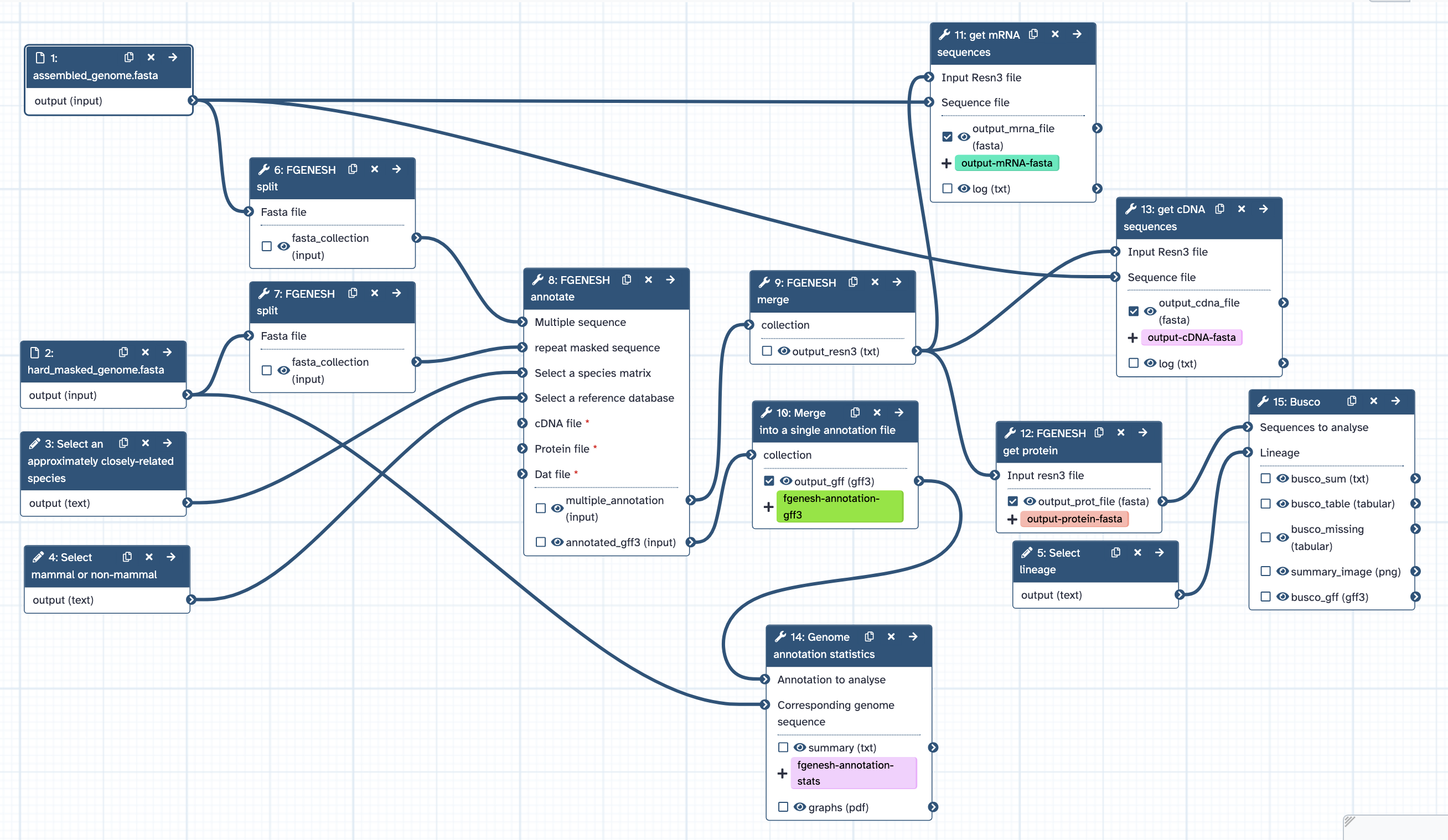
Fig 1. Genome annotation workflow diagram.
Further to this, a summary of the different elements of this assembly approach are detailed below:
| Process name | Workflow name | Description | Inputs | Outputs |
|---|---|---|---|---|
| UPLOAD FILES | Not applicable | See the different upload options. | reference genome, repeat masked reference genome, .cdna, .pro and .dat files from transcriptome assembly workflow |
Uploaded data! |
| GENOME ANNOTATION | Fgenesh annotation-TSI | Splitting of reference and masked reference genome files, annotation of genome using FgenesH++, merging of annotation files and extraction of mRNA, CDS and protein sequences | reference genome, repeat masked reference genome, .cdna, .pro and .dat files from transcriptome assembly workflow |
GFF3 of annotated genes, fasta file of mRNA, CDS and protein sequences which were annotated |
In-depth workflow guide
Register and login
- To register for Galaxy Australia, visit the login page.
- Click the
Register herelink, as shown in Fig 2. - Complete the registration wizard and click
Create. - Login to your account!
Fig 2. Log-in / registration menu for Galaxy Australia.
Upload data file(s)
- In Galaxy Australia, create a new history and click on
Upload Data - Choose local files (see Figure 3)
Fig 3.
- Upload your assembled reference genome and masked reference genome (Link to repeat masking workflow), as well as the
.cdna,.proand.datoutput from your transcriptome assembly. Note: it is recommended by Softberry that the genome is hard-masked rather than soft-masked.
Run the annotation workflow
- Make sure you are logged into Galaxy Australia
- Visit this link to:
- retrieve the workflow for genome annotation, and
- import into your Galaxy Australia workflows
- Once you have reached the workflow screen, select the
playbutton for the annotation workflow (See Figure 4)
Fig 4.
-
The workflow invocation window will open.
-
Select your reference assembled genome fasta file.
-
Select your (hard) repeat-masked reference genome fasta file.
- Select the matrix of an approximately closely-related species (full list of matrices).
- Select “mammal” or “non-mammal” database.
- Select a lineage that BUSCO will use on the output protein file.
- For “Select nr db type”, note that this only applies if you are using a set of known protein sequences, and have selected the option further down: USE_PROTEINS = yes. In this case, select the appropriate NR database (e.g. TSI animals). If you aren’t using proteins, disregard this and leave as default setting.
- Select the correct inputs for cDNA, protein and dat files.
- Click “Run Workflow”.



School of Life and Environmental Sciences, The University of Sydney, NSW, Australia