Galaxy Australia is capable of performing genome repeat masking and transcriptome alignment with stringtie and transdecoder
This How-to-Guide will describe the steps required to repeat your genome and align transcripts on the Galaxy Australia platform (see Fig 1), developed in consultations between the Bioplatforms Australia Threatened Species Initiative, Galaxy Australia, and the Australian BioCommons.
If you need help, the Galaxy community is both approachable and helpful. Ask them questions!
Quick start guide
- Login to Galaxy Australia
- Create a new history
- Upload your assembled reference genome and raw RNA sequencing reads (can be imported from Bioplatforms Australia Portal)
- Load and execute workflows, using required options
- Review workflow report and perform additional QC as needed
- Re-run workflows, or individual tools, as needed
How to cite the workflows
Silver, L., & Syme, A. (2024). Repeat masking - TSI. WorkflowHub. https://doi.org/10.48546/WORKFLOWHUB.WORKFLOW.875.3
Silver, L., & Syme, A. (2024). QC and trimming of RNAseq reads - TSI. WorkflowHub. https://doi.org/10.48546/WORKFLOWHUB.WORKFLOW.876.1
Silver, L., & Syme, A. (2024). Find transcripts - TSI. WorkflowHub. https://doi.org/10.48546/WORKFLOWHUB.WORKFLOW.877.1
Silver, L., & Syme, A. (2024). Combine transcripts - TSI. WorkflowHub. https://doi.org/10.48546/WORKFLOWHUB.WORKFLOW.878.1
Silver, L., & Syme, A. (2024). Extract transcripts - TSI. WorkflowHub. https://doi.org/10.48546/WORKFLOWHUB.WORKFLOW.879.1
Silver, L., & Syme, A. (2024). Convert formats - TSI. WorkflowHub. https://doi.org/10.48546/WORKFLOWHUB.WORKFLOW.880.1
The overall workflow
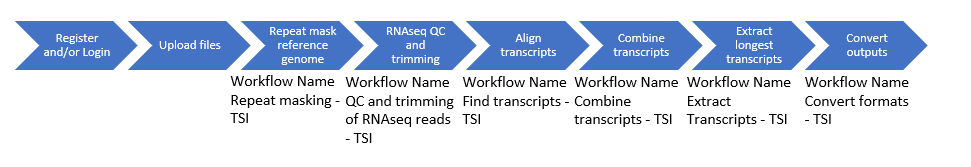
Fig 1. The overall transcriptome alignment workflow.
Further to this, a summary of the different elements of this alignment approach are detailed below:
| Process name | Workflow name | Description | Inputs | Outputs |
|---|---|---|---|---|
| UPLOAD FILES | Not applicable | See the different upload options. | reference genome, Fastq mRNA | Uploaded data! |
| Repeat Masking | Repeat masking - TSI | Repeat masking of reference genome | Reference genome | FASTA files of hard-masked and soft-masked genomes, Statistic file |
| RNA seq QC and trimming | QC and trimming of RNAseq reads -TSI | Trimming of fastq files, including a fastqc step | Raw mRNA sequencing files | FASTQC report, Paired read FASTQ file |
| Align reads to find transcripts | Find transcripts - TSI | Alignment of trimmed FASTQ reads to masked reference genome | (soft) repeat masked reference genome, paired trimmed FASTQ reads | BAM file, GTF file alignment metrics |
| Combine Transcripts | Combine Transcripts - TSI | Merges individual tissue transcripts to a global transcriptome and predicts coding sequences | GTF file, soft-masked genome, closely related species coding and non-coding sequences | GTF for global transcriptome, FASTA sequences of coding transcripts |
| Extract Longest Transcripts | Extract Transcripts-TSI | Transdecoder predictions and filtering of transcripts | FASTA sequence of coding transcripts | pep.fasta, cds.fasta and gff3 file of longest isoform transcripts |
| Convert Outputs | Convert formats - TSI | Converts outputs of transcdecoder to required inputs for FGenesH++ annotation | transdecoder-peptides.fasta, global_nucleotides.fasta | .cdna, .dat and .pro files |
In-depth workflow guide
Register and login
- To register for Galaxy Australia, visit the login page.
- Click the
Register herelink, as shown in Fig 2. - Complete the registration wizard and click
Create. - Login to your account!
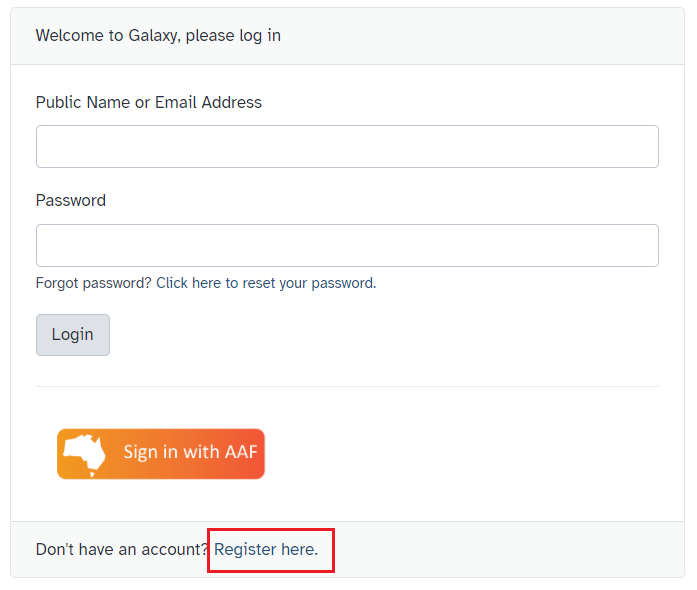
Fig 2. Log-in / registration menu for Galaxy Australia.
Upload data file(s)
- In Galaxy Australia, create a new history and click on
Upload Dataand choose local files (See Figure 3) - Upload your assembled reference genome and raw mRNA transcriptome FASTQ files
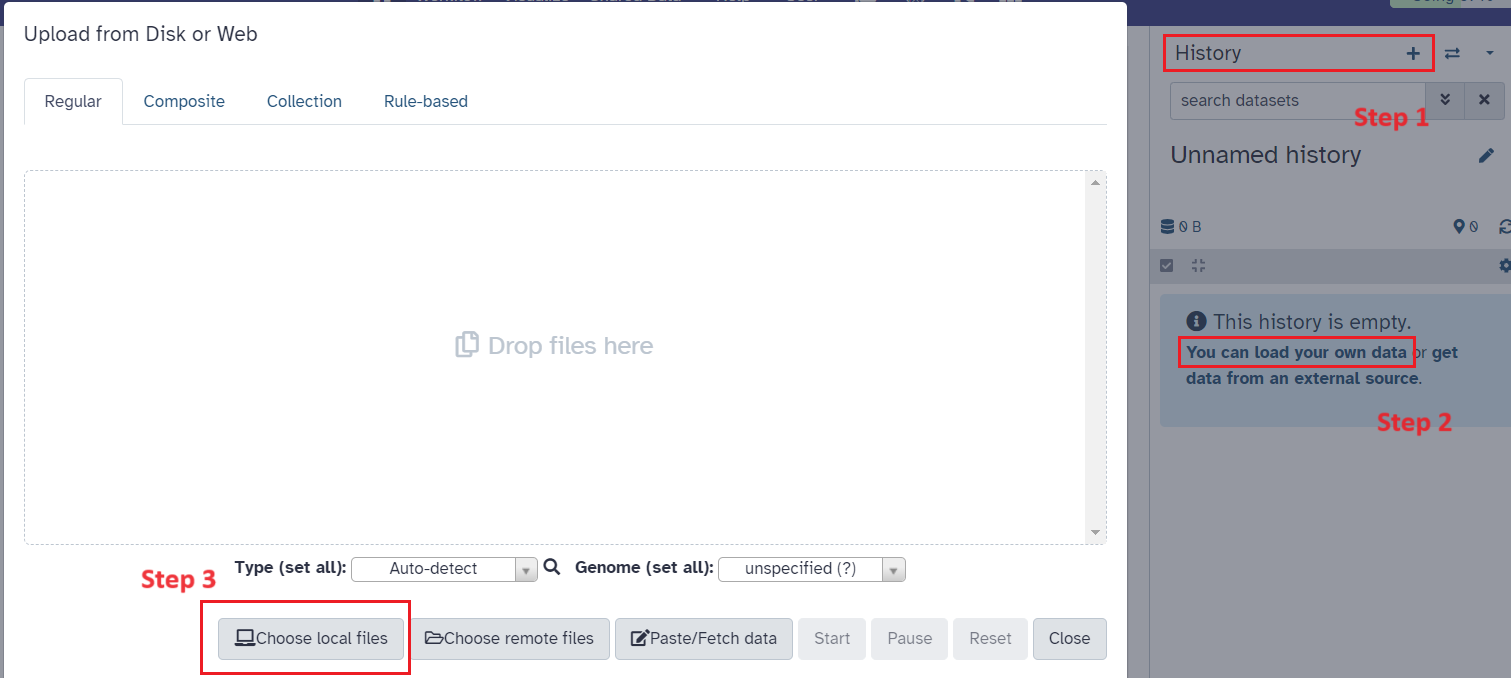
Fig 3.
Run the Repeat Masking Workflow
- Make sure you are logged into Galaxy Australia
- Visit this link to:
- Retrieve the workflows for
Repeat Masking - Import into your Galaxy Australia workflows
- Retrieve the workflows for
- Once you have reached the workflow screen, select the
playbutton for Repeat Masking (See Figure 4) - The workflow invoation window will open. Select your reference genome fasta file (Figure 5) and run workflow
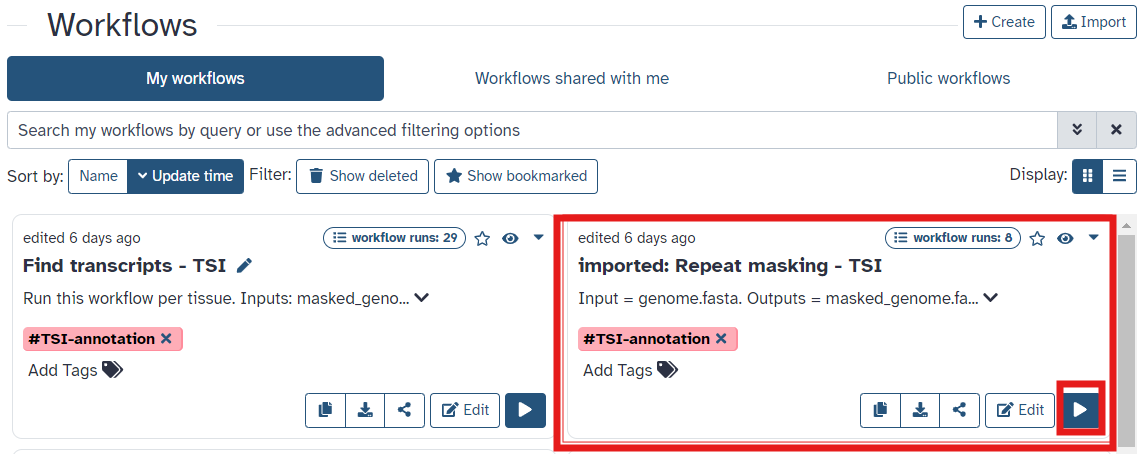
Fig 4.
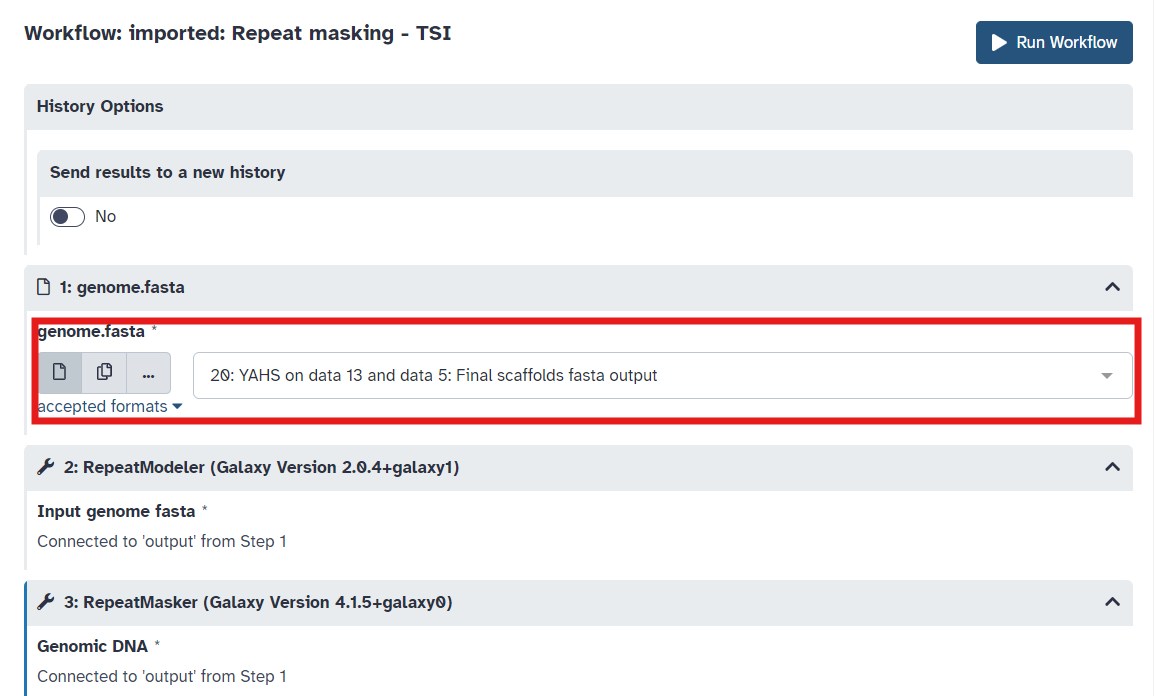
Fig 5.
Run RNA seq QC and trimming workflow
- Visit this link to:
- Retrieve the workflows for
QC and trimming of RNAseq reads - Import into your Galaxy Australia workflows
- Retrieve the workflows for
- If each tissue was sequences across multiple lanes, make two datasets, one containing forward reads and one containing reverse reads
- Once you have reached the workflow screen, select the
playbutton for QC and trimming of RNAseq reads (See Figure 6). - Select forward and reverse datasets and run workflow (Figure 7)
- Check the MultiQC webpage for each trimmed collection and “#table-reads-info-after-trimming” files to quality check trimmed reads
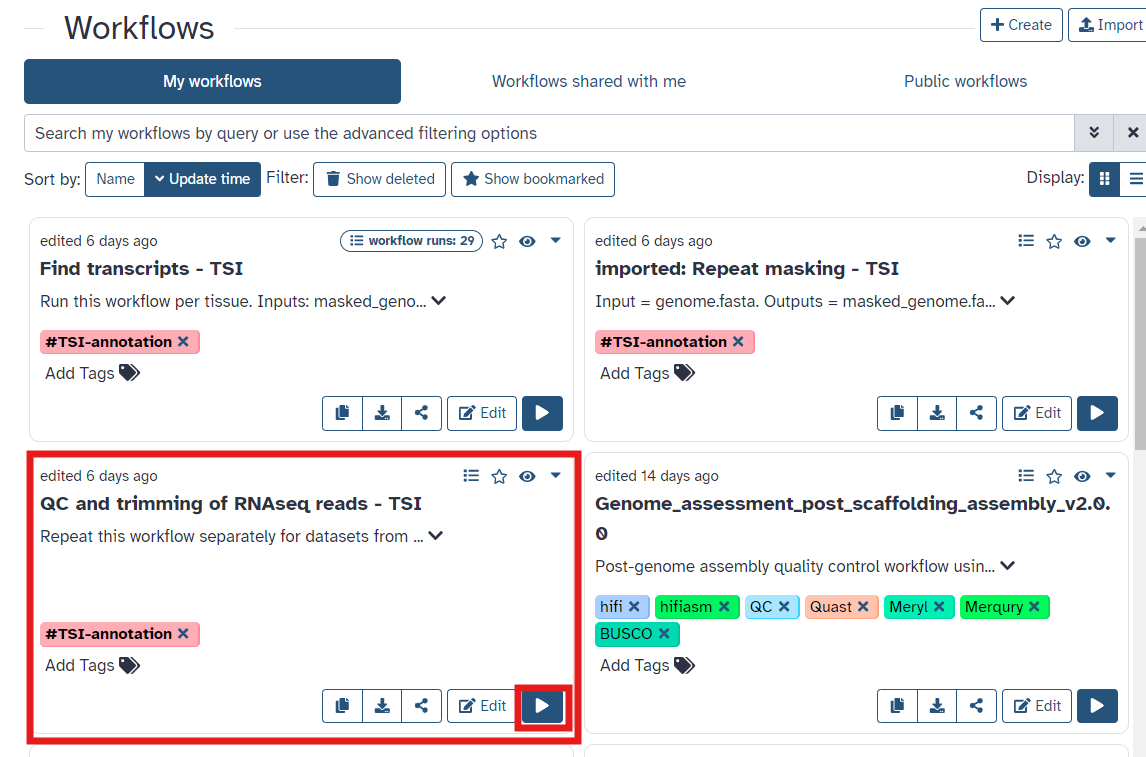
Fig 6.
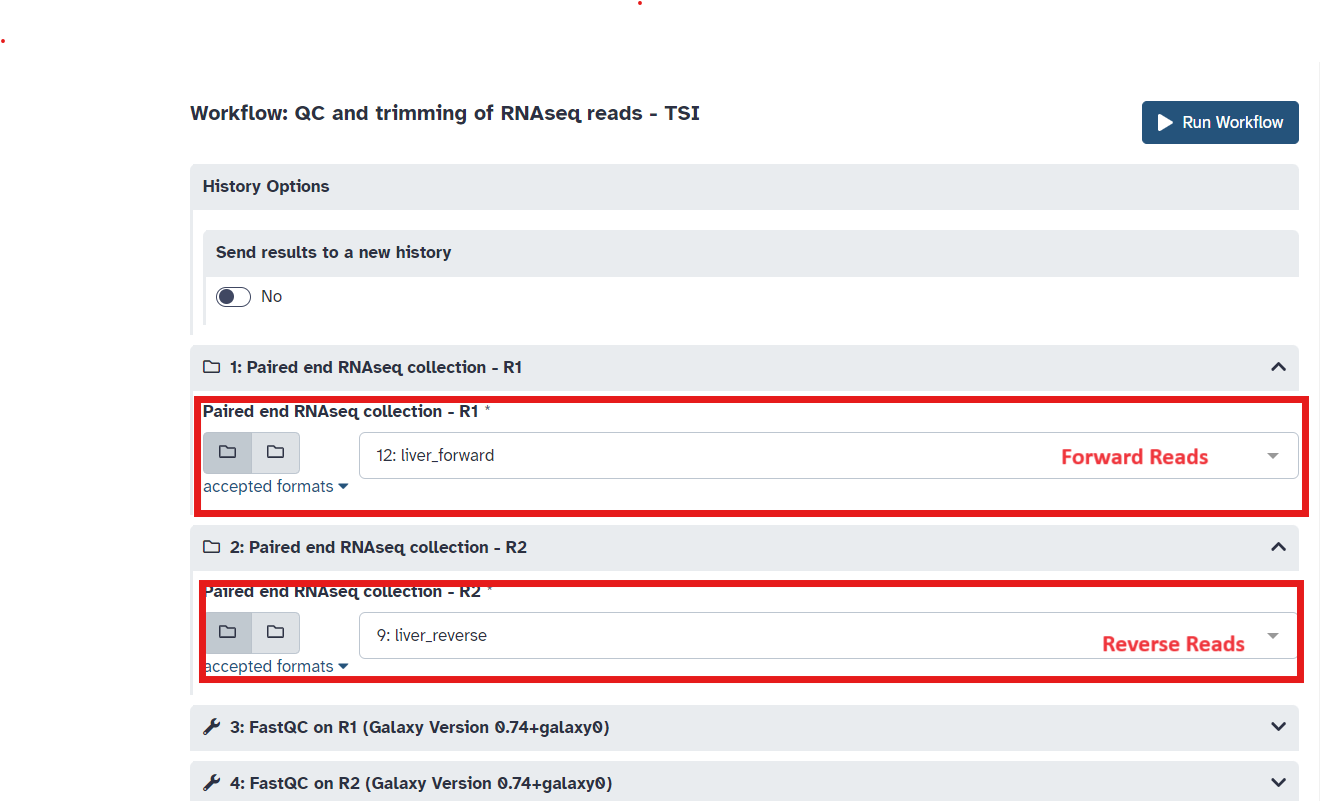
Fig 7.
Run align reads to find transcripts workflow
- Visit this link to:
- Retrieve the workflows for
Align reads to find transcripts - Import into your Galaxy Australia workflows
- Retrieve the workflows for
- Once you have reached the workflow screen, select the
playbutton for Align reads to find transcripts (Fig 8) - Select the paired forward and paired reverse trimmed reads and soft-masked reference genome as input (Fig9), ensure you select files tagged with
#fastq_out_r1_pairedand#fastq_out_r2_paired - Check the mapping summary file for each tissue to make sure there are high mapping rates to the genome
- Make a dataset collection containing gtf files for all tissue transcriptomes
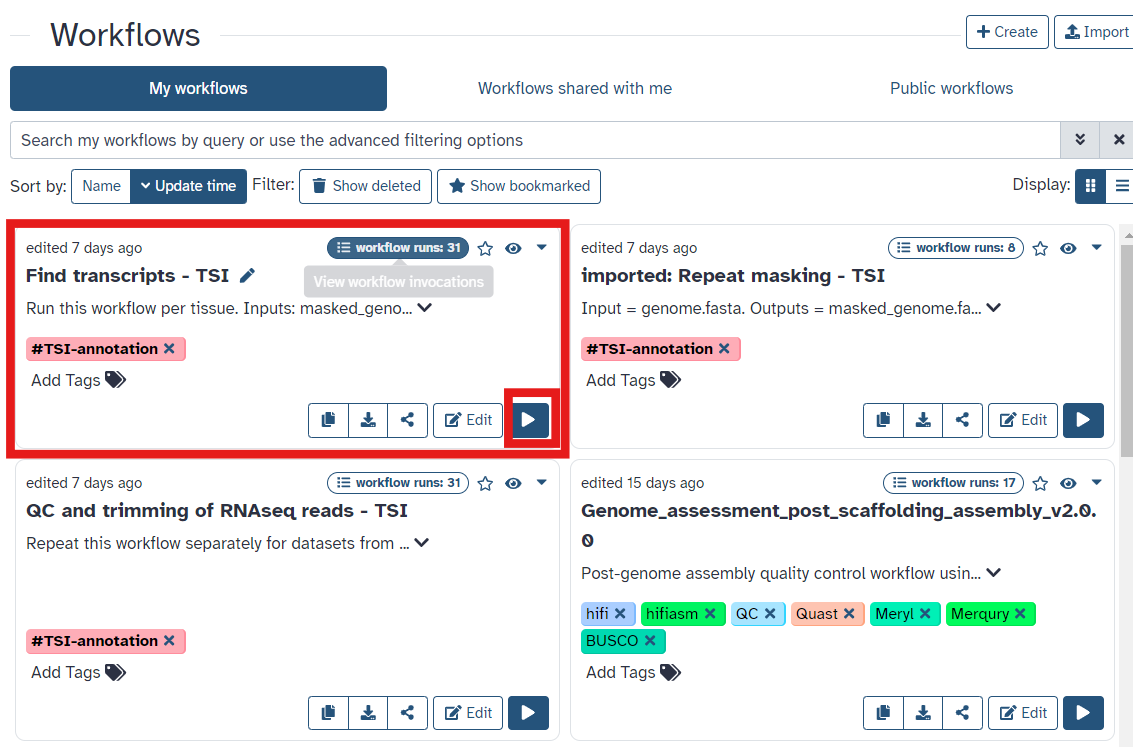
Fig 8.
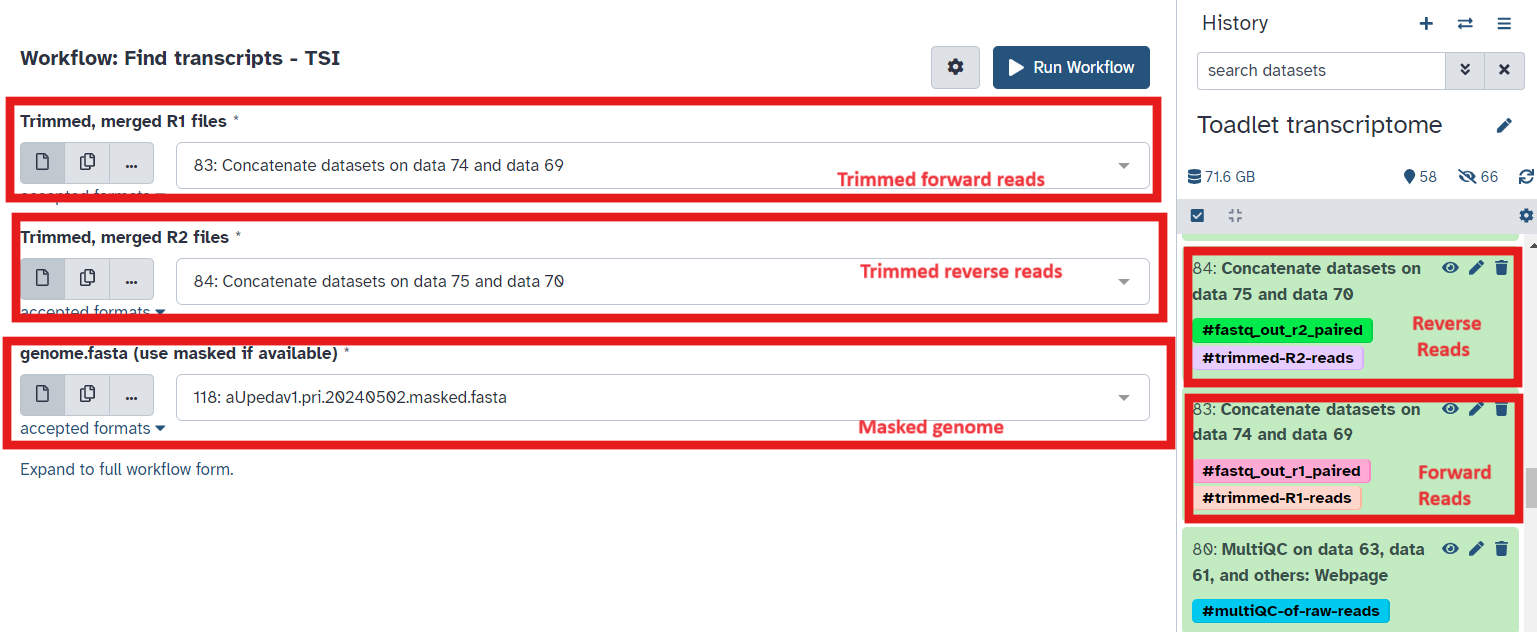
Fig 9.
Run Combine Transcripts
- Visit this link to:
- Retrieve the workflow for
Combine transcripts - Import into your Galaxy Australia workflows
- Retrieve the workflow for
- Once you have reached the workflow screen, select the
playbutton for Combine Transcripts (Fig 10)
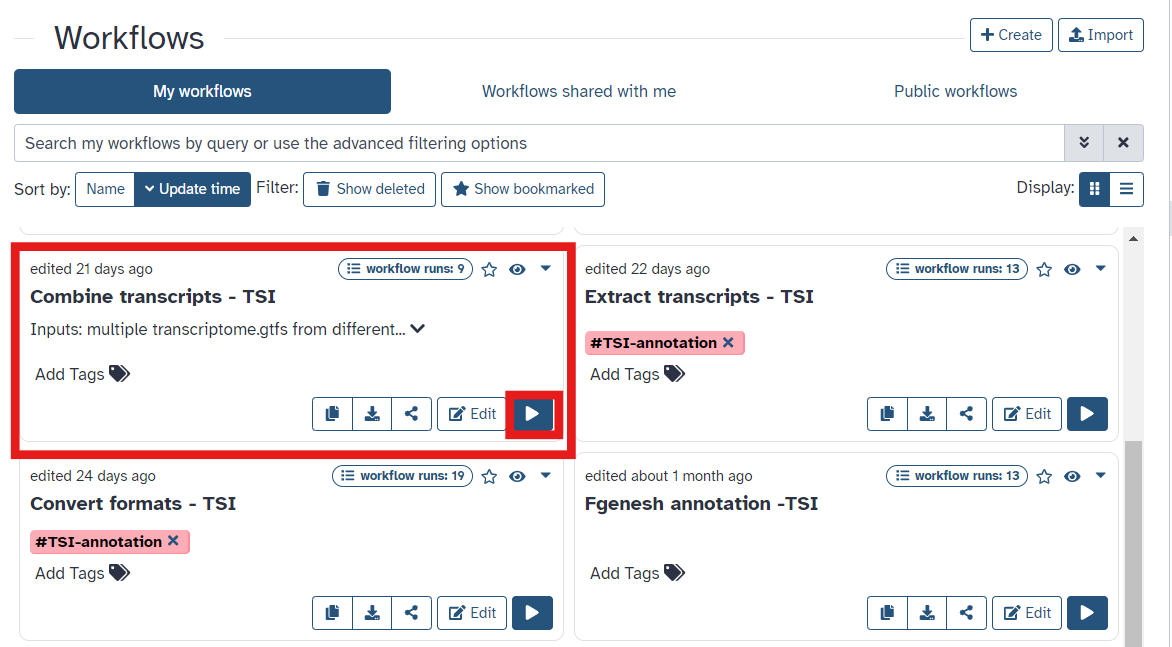
Fig 10.
- Search for your species on NCBI Taxonomy to find the most closely related species which has an NCBI RefSeq annotation (Fig 11)
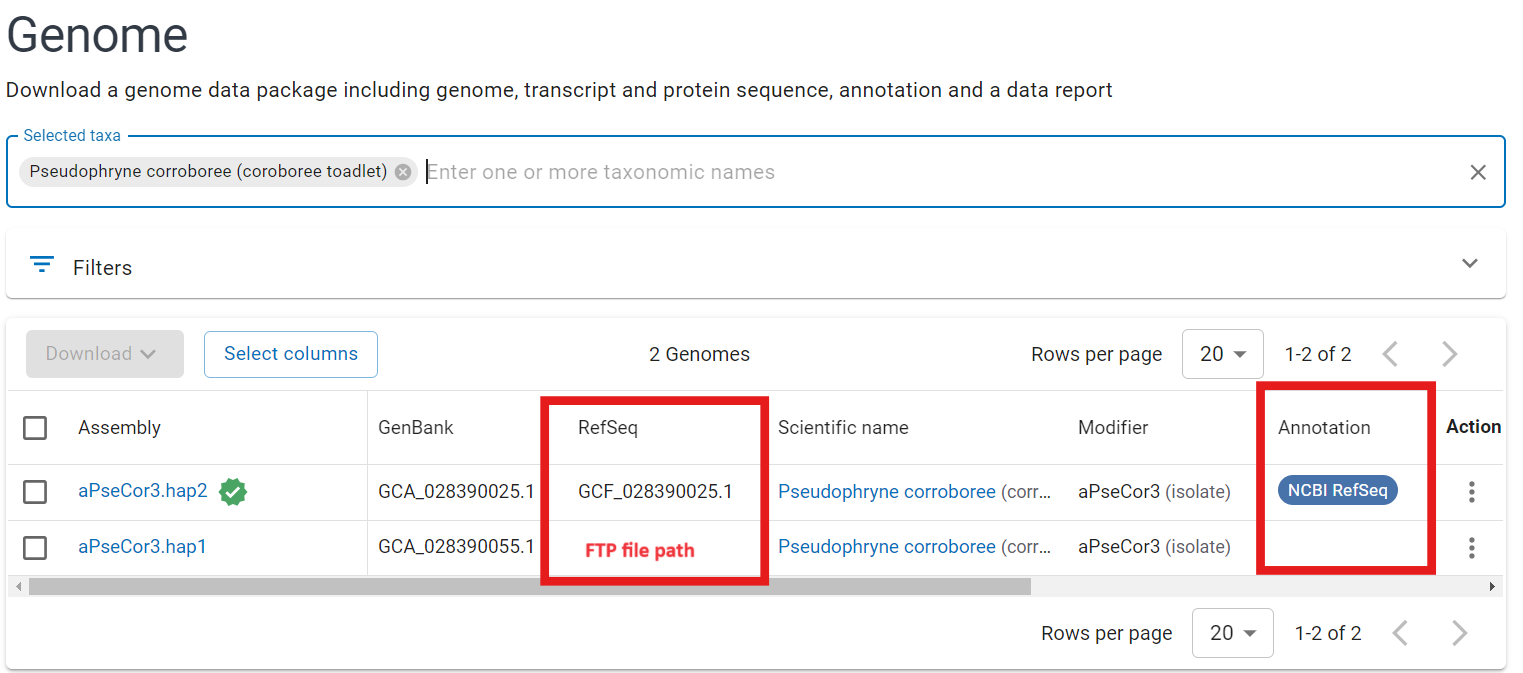
Fig 11.
- Go to the NCBI ftp server and locate the entry for this species (e.g. Corroborree frog RefSeq entry is GCF_028390025.1 and ftp entry is https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/028/390/025/)
- Download the
_cds_from_genomic.fna.gzandpseudo_without_product.fna.gzfiles to your local computer and upload into Galaxy (Fig 12)
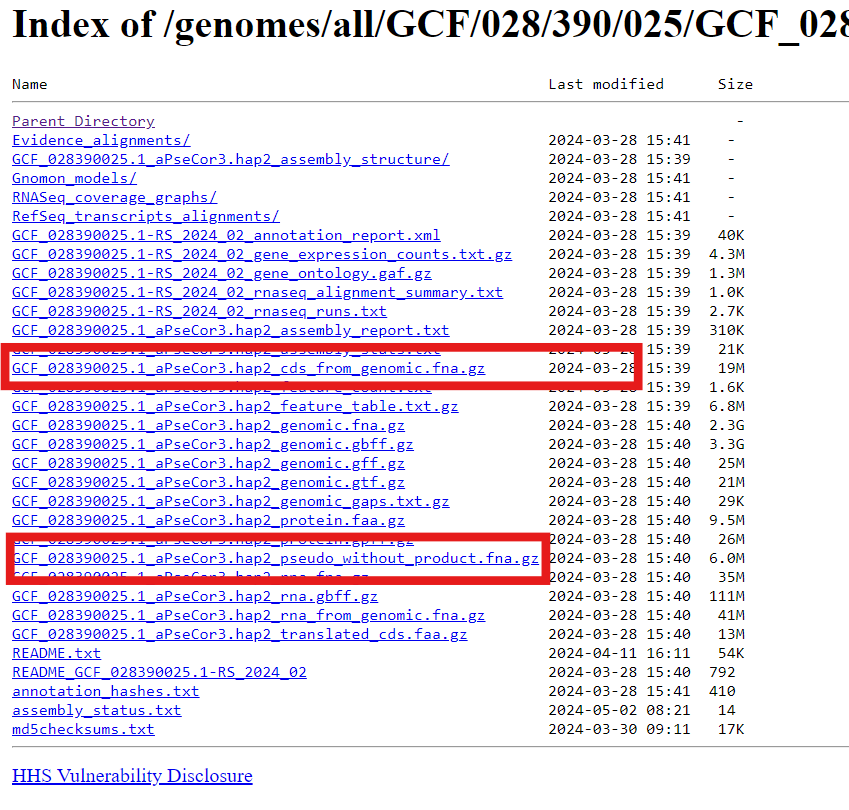
Fig 12.
- Select the gtf collection, soft-masked reference genome, coding sequences and pseudo coding sequences in the combine transcripts workflow
- In Step 7 of the workflow ensure the masked genome is selected and that in Step 10 of the workflow type “1” in the
List of Fieldsbox (Fig 13; Fig 14; Fig 15)
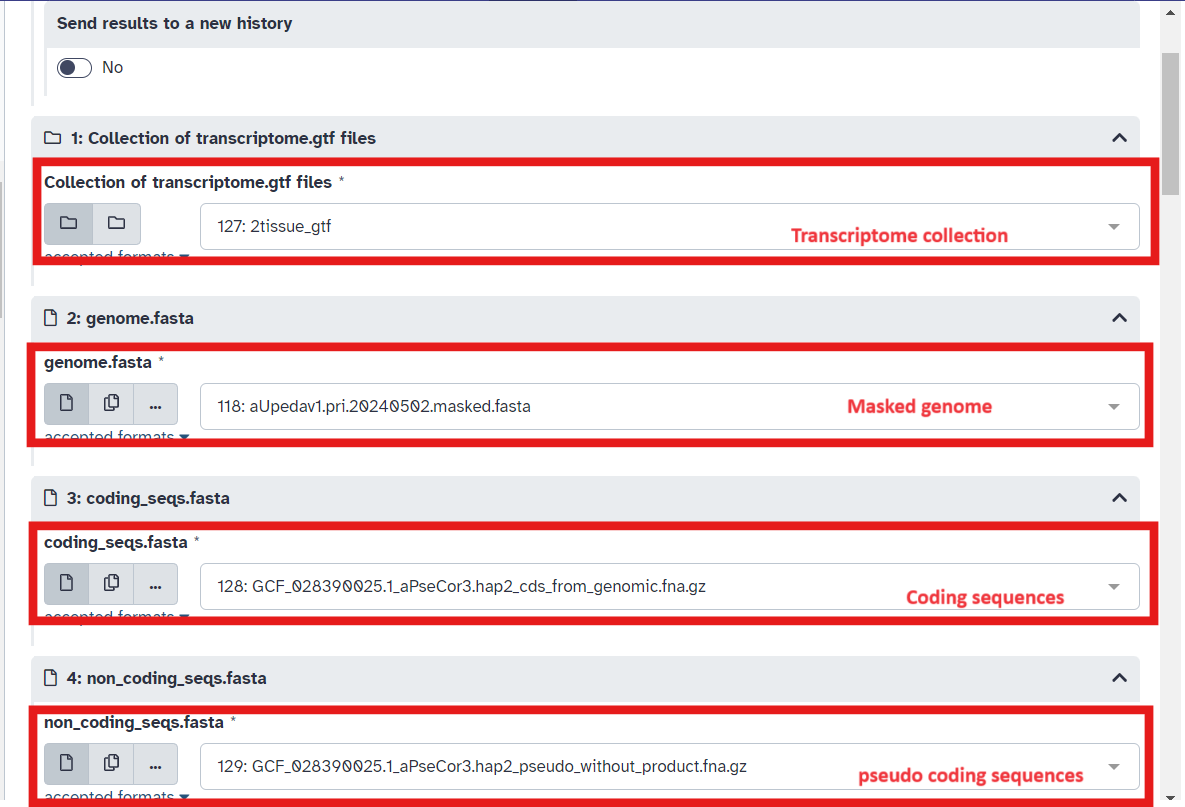
Fig 13.
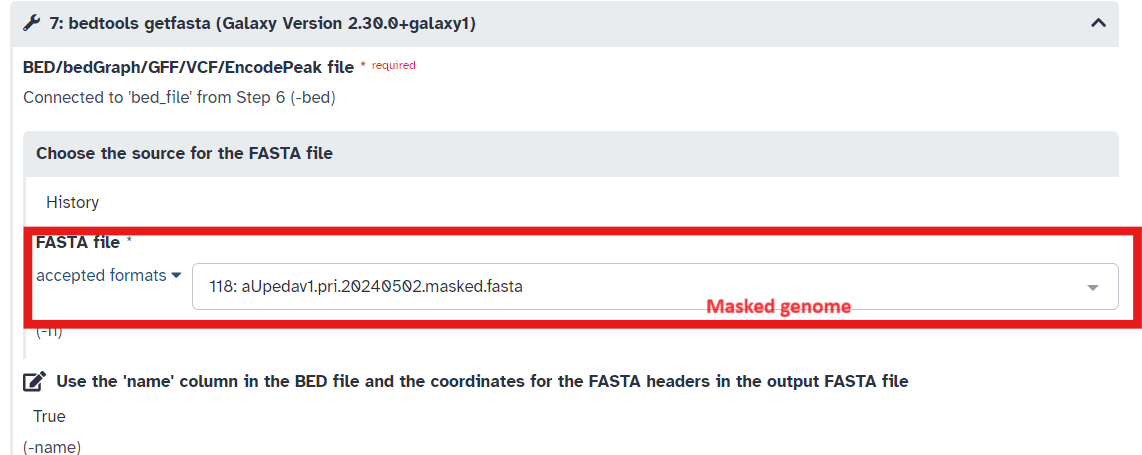
Fig 14.
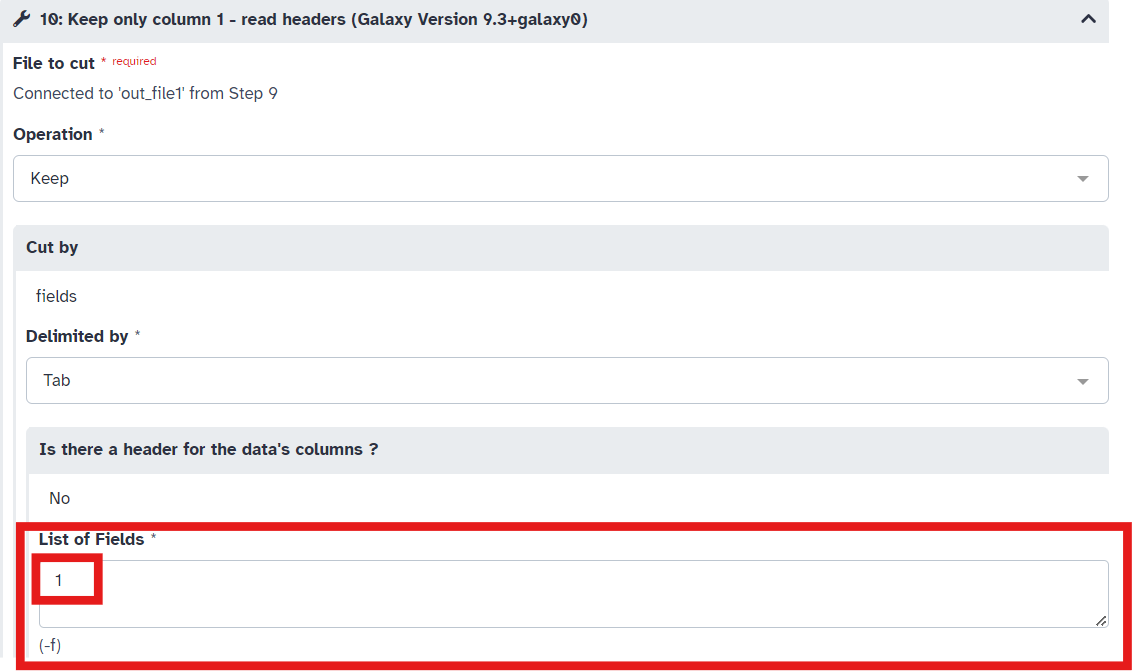
Fig 15.
Run Extract Longest Transcripts workflow
- Visit this link to:
- Retrieve the workflows for
Extract longest transcripts - Import into your Galaxy Australia workflows
- Retrieve the workflows for
- Once you have reached the workflow screen, select the
playbutton forExtract longest transcript(Fig 16)
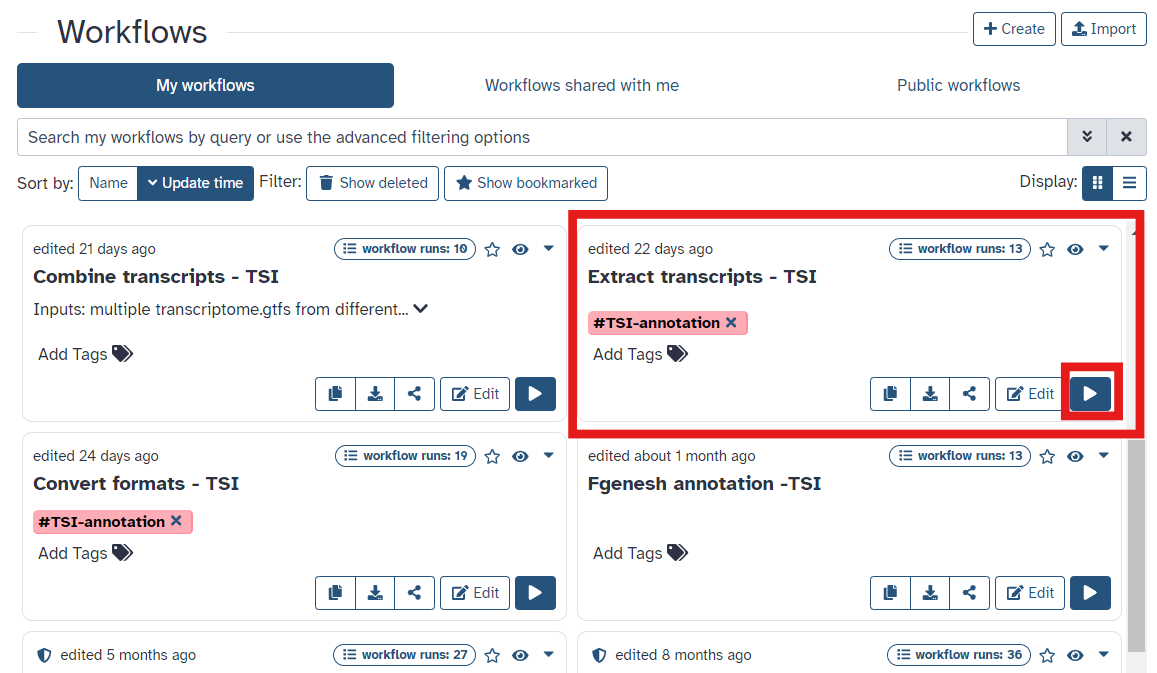
Fig 16.
- Select the fasta output file from the previous workflow (tagged with #seqs-with-high-coding-prob) as input to the workflow (Fig 17)
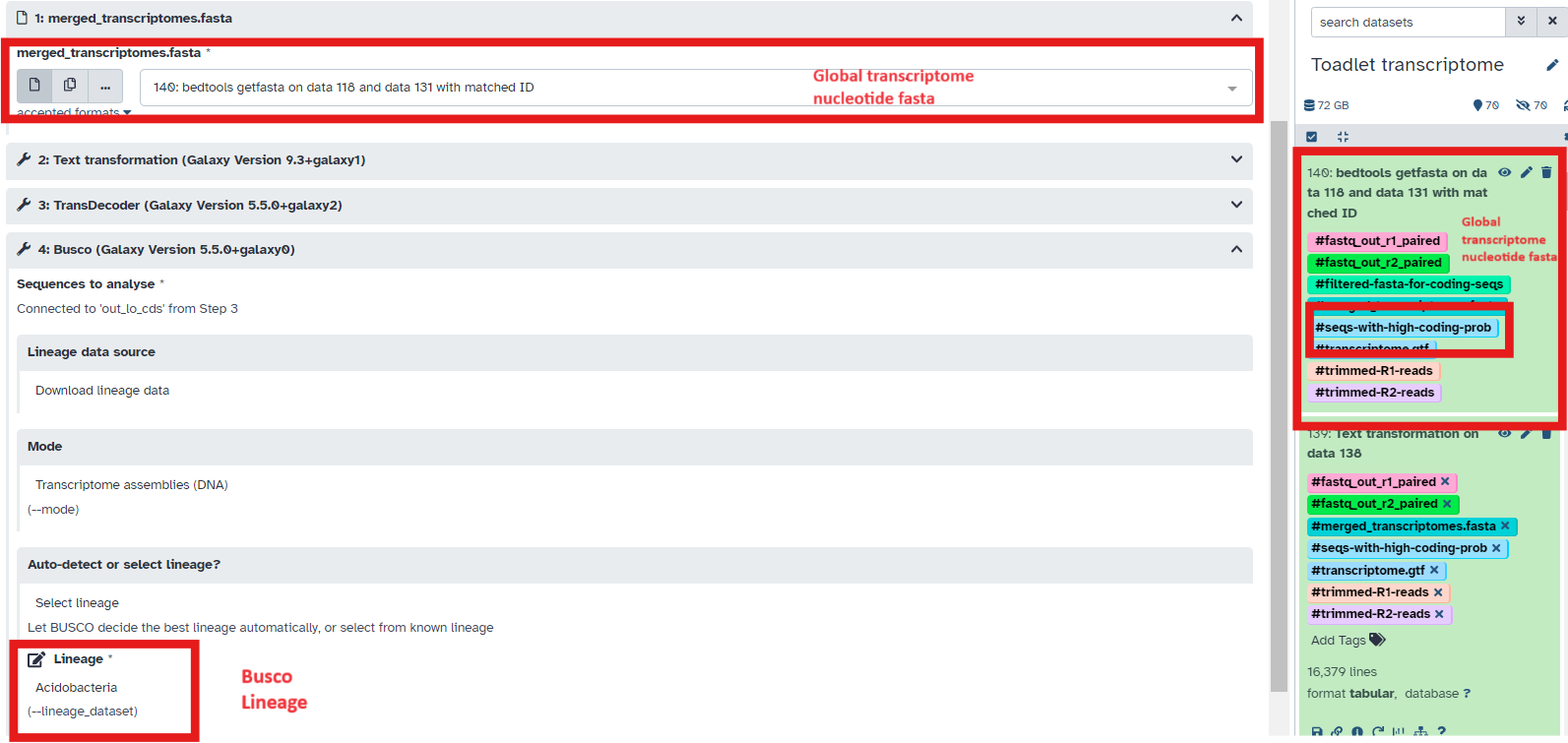
Fig 17.
- Also, in workflow step 4, select the most appropriate BUSCO lineage to run on the output file from Transdecoder
- Check the BUSCO output to ensure a high percentage of complete BUSCO’s are found in the transcriptome
Run Convert Outputs workflow
- Visit this link to:
- Retrieve the workflows for
Convert outputs - Import into your Galaxy Australia workflows
- Retrieve the workflows for
- Once you have reached the workflow screen, select the
playbutton for Covert outputs (Fig 18)
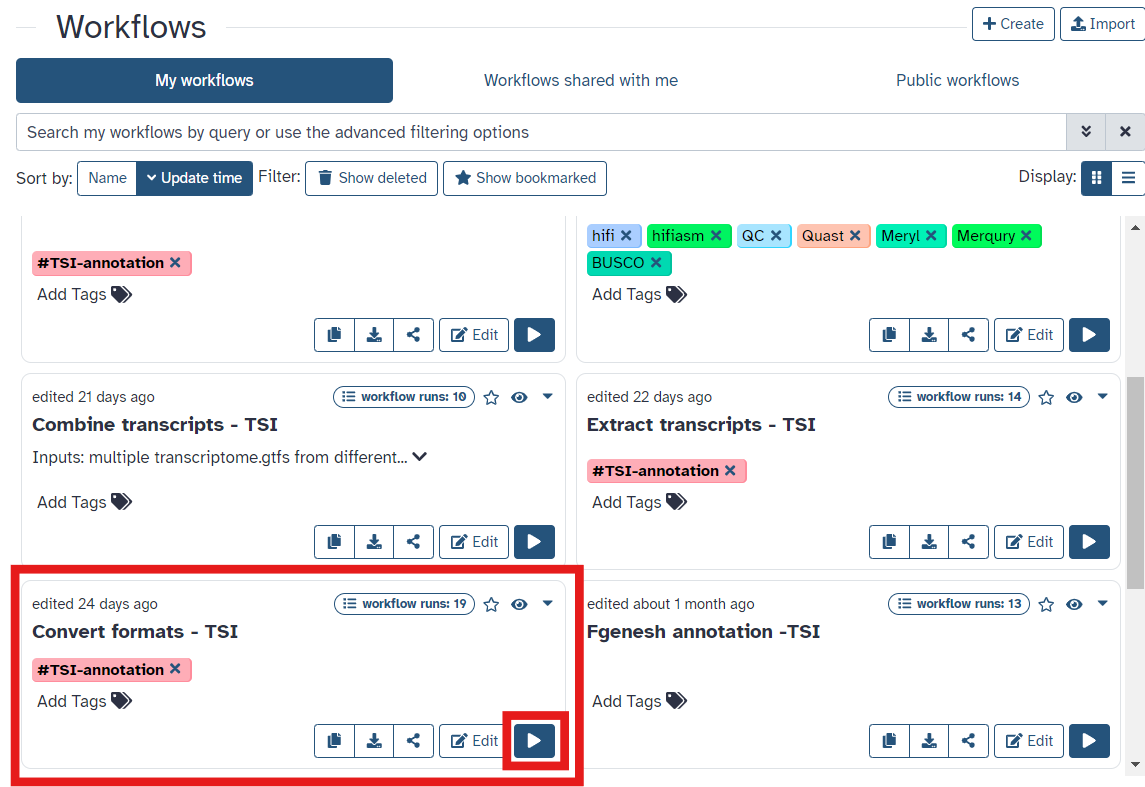
Fig 18.
- Select the transdecoder peptide fasta file and the text transformed fasta output file from the Combine Transcripts workflow (Fig 19)
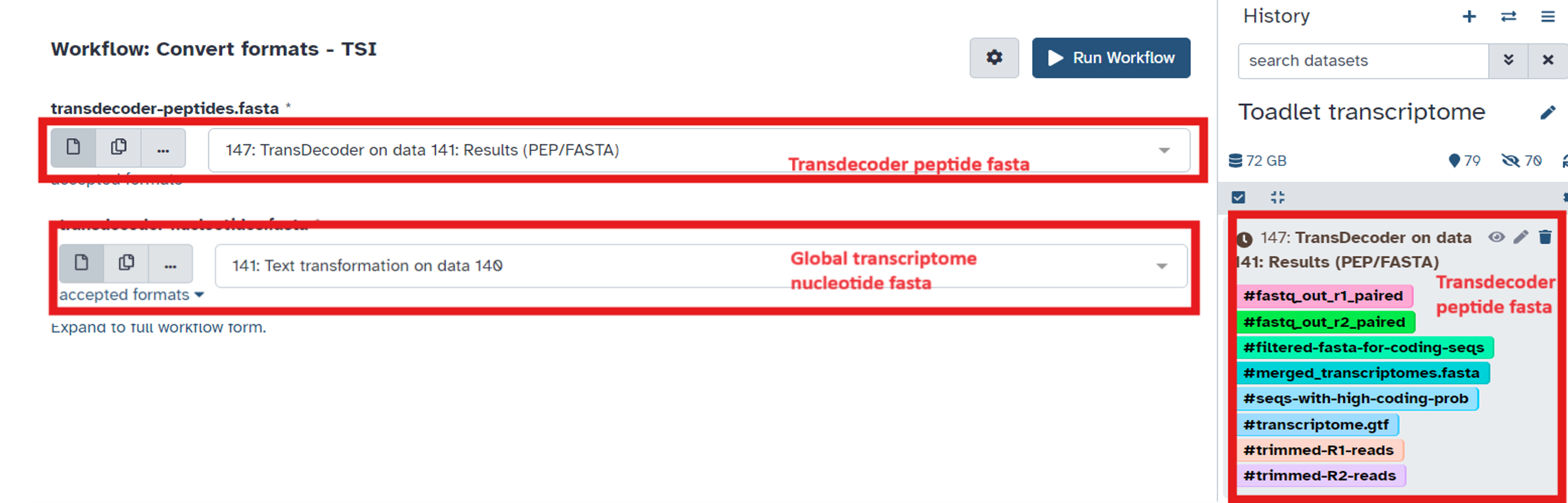
Fig 19.
- The output files tagged with
#dat,#pro, and#cdna, along with the hard-masked and unmasked reference genome are used as input files for FGenesH++ genome annotation.



School of Life and Environmental Sciences, The University of Sydney, NSW, Australia